作者:炸鸡
上一期《走进消除反应Elimination Reactions(1)——怎样让消除反应发生?》笔者谈到有些卤代烃是既可以发生消除反应也可以发生取代反应,通过选择不同的亲核试剂或调节温度可以使得消除反应或取代反应成为主反应。这一期笔者将把重心转移到消除反应上,探讨不同的消除反应机理之间有哪些差别。
E1和E2机理概述
在亲核取代反应中,根据反应机理的不同可以分为SN1反应和SN2反应。类似的,消除反应根据反应机理的不同分为E1反应和E2反应。E是Elimination Reactions的首字母大写,E1和E2里的1和2则寓意着各自反应机理的差别。
E1中的1指代这个反应机理是单分子反应机理。反应机理:离去基团离去,然后β位的质子被脱掉,形成双键,产物生成(图1)。

图1. E1机理
E2中的2指代这个反应机理是双分子反应机理。反应机理:亲核试剂(在E2里更具体一点为碱)夺取β位的质子和离去基团离去同时发生,形成双键,产物生成(图2)。

图2. E2机理
在E1机理的反应中,离去基团离去后,原来的反应物变成了一个含碳正离子(carbocation)的化合物,碳正离子非常以及极其活泼,碳正离子就好像反应溶液中的“躁动分子”,只要它一旦生成,它就会不管三七二十一立即与它周围任何可以反应的物质发生反应或是迅速发生重排。所以对于E1机理的反应来说,只要碳正离子生成,反应几乎是瞬间结束的(图3),所以决定E1机理的反应的速度的只有离去基团。离去基团离去的慢,反应速度就慢;反之,离去基团离去地快,反应速度就快。
Rate(E1)=k[RX]

图3
在E2机理的反应中,亲核试剂(对E2里更具体一点为碱)的进攻和离去基团的离去是同时发生的,不存在碳正离子。如果亲核试剂是个强碱或离去基团离去的很快,那么反应速度就会变快。所以决定E2机理的反应的反应速度有离去基团和亲核试剂两个因素。
Rate(E2)=k[RX][base]
哪些条件会促成E1机理
①如上文所讲,碳正离子的生成对E1机理的反应至关重要。只要能生成相对稳定的碳正离子,那么反应机理就倾向于变成E1。那么什么样的碳正离子才算是稳定的呢?这要从碳正离子的结构说起:一般的饱和碳是sp3杂化,碳正离子的碳是sp2杂化,空的p轨道垂直于其他C-H键所在的平面。sp2杂化意味着不再是“金字塔”状的共价键排布,而是“螺旋桨”状的共价键排布(三个桨叶共平面)(图4)。

图4. 饱和碳(右)正离子碳(左)
碳正离子的空的p轨道需要临近的其他基团向它“输送”电子,让碳正离子的空的p轨道不那么“缺电子”从而变得更稳定。
a)与烷基相连的碳正离子会比较稳定。例如图5所示,碳正离子与三个甲基相连,每个甲基上总有一个C-H键能大致与碳正离子的p轨道平行,能与碳正离子的p轨道平行的那个C-H键上的σ电子会“涌入”p轨道,进而稳定碳正离子(图5)。

图5. 一个稳定的碳正离子
b) 如果碳正离子的ɑ位有可以让碳正离子的p轨道离域的π电子(图6),碳正离子也会得到稳定。此时三个p轨道(两个满,一个空)组成了一个分子轨道,新组成的分子轨道相较原本的原子轨道拥有更低能量的HOMO,也就更稳定了。

图6
c) 有富电子的芳香环(对位或邻位有给电子基)与碳正离子的p轨道形成共轭,那么碳正离子也会稳定。
d) 如果碳正离子与有孤对电子的杂原子相连,孤对电子会进入碳正离子空的p轨道,稳定碳正离子。即使碳正离子是伯碳,也很稳定(图7)。

图7
下面是几例稳定的碳正离子和不稳定的碳正离子(图8):

图8
能够形成稳定碳正离子的底物被认为是利于E1机理发生。
②除了产物的结构可以稳定碳正离子外,反应溶剂的选择也十分重要。极性溶剂是利于E1机理发生的,因为极性溶剂能很好地稳定碳正离子。
③E1反应速率与离去基团有关(Rate(E1)=k[RX])。如果离去基团的离去能力很好再加上弱碱甚至没有碱,E1反应会发生。
④最后一个最简单——酸性条件。这很容易理解,如果离去基团是-OH(消除反应里的离去基团除了卤素原子外还有很多),在碱性条件下-OH是永远没法离去的,只有在酸性条件下被质子化形成-O+H2后离去。既然反应条件为酸性,E2机理是绝对不可能了因为E2反应的发生需要碱的进攻和离去基团同时发生。所以酸性条件下的消除反应的机理通通为E1机理。(图9)

图9
哪些条件会促成E2机理
E2反应的反应速率=k[RX][base]。要想E2反应发生,必须确保亲核试剂(在E2里具体为碱)有足够强的能力夺取质子且离去基团又足够快速地离去。所以使用强碱+强离去基团的组合利于E2反应的发生。和亲核取代不同的是,E2反应的发生不限定于特定的底物结构,许多可以发生E1反应的化合物只要加以强碱一样可以变为E2反应。
注意:E1,E2和SN1,SN2之间没有一点关系
有一些同学会把E1,E2和SN1,SN2之间强行关联起来,认为可以发生SN1的底物会发生E1消除反应,认为可以发生SN2的底物会发生E2消除反应。这是个误区,E1,E2和SN1,SN2之间没有一点关系。反例如下(图10):

图10
氯代叔丁烷既可以发生E1消除反应,也可以发生E2消除反应(取决于碱的强弱),但由于氯代叔丁烷ɑ-碳的周围有太多甲基造成位阻太大,所以只能发生SN1亲核取代反应。
E1cB机理
当离去基团先离去,碱夺质子很慢,反应机理为E1;当离去基团和碱夺质子同时发生时,反应机理为E2。那么,如果当碱的碱性很强以至于先夺取了质子,离去基团再离去会发生什么呢?——这就是E1cB机理(图11)。

图11. E1cB机理

图12. E1,E2,E1cB三者之间的关系(图片改编自[2])
当强碱先夺取β位的质子后,碳负离子生成,而后碳负离子迫使离去基团离去。当β位碳负离子很稳定(旁边有吸电子基如硝基羰基)且碱为强碱(如果碱的碱性不足以脱去β位的质子,E1cB反应就不会发生)时,反应机理偏向于E1cB机理。
Rate(E1cB)=k[RX][base]

图13. 一个E1cB机理的反应
学有余力
我们所说的E1机理,E2机理,E1cB机理描述的都是一种极端的情况,在现实中的消除反应中,反应的机理多是介于极端机理的中间机理,例如现实中用的最多的是“偏向于”“倾向于”。例如这个反应机理偏E1机理,或这个反应机理位于或处于E1机理和E2机理之间。

图14(图片改编自[2])
参考文献
[1] J. Clayden, N. Greeves, S. Warren, 2012. Organic Chemistry. Great Clarendon Street, Oxford: Oxford University Press. 1187pp. 2nd edition. [2] F. A. Carey, R.J. Sundberg, Advanced Organic Chemistry Part A: Structure and Mechanisms.封面图片来自:Image by <a href=”https://www.freepik.com/free-vector/science-lab-with-objects_7405691.htm#query=chemical%20reaction&position=7&from_view=keyword&track=ais&uuid=c9ebc29e-3e39-4c5d-8000-993a1cf63f69″>Freepik</a>
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载.
概要
Nozaki-Yamamoto消除(Nozaki-Yamamoto elimination)是在氨基有机铝(organoaluminum amide, 又称为铝氨基碱,aluminum amide base)试剂 2,2,6,6-四甲基哌啶基二乙基铝 (diethylaluminum 2,2,6,6-tetramethylpiperidide, DATMP)[1]-[3]作用下,环氧化物进行的高度区域及立体专一性开环重排过程,生成相应烯丙醇类化合物的反应[1]-[3]。

该反应在1974年由日本京都大学工业化学系 (京都大学工学部工業化学教室, Department of Industrial Chemistry, Kyoto University)的山本尚 (山本 尚, Yamamoto Hisashi)与野崎一 (野崎 一, Nozaki Hitoshi)教授首次报道[1]。Nozaki-Yamamoto消除反应极高的区域及立体专一性源于铝氨基碱试剂比之前报道的锂氨基碱[4]-[6]与丁基锂[7]试剂具有更高的立体位阻,以及该试剂特有的Lewis酸-碱协同反应 (LA-LB cooperative reaction)特性。反应完成后,手性环氧化物的构型保持,同时形成trans-双键。该反应对于与碱存在竞争性亲核加成反应的末端环氧化物同样适用。1977年该研究组采用N-甲基苯胺基二乙基铝,将底物扩展至氧杂环丁烷,成功实现了高烯丙醇的合成[8]。

目前已经广泛应用于各类烯丙醇[1]-[3]及高烯丙醇[8]类化合物的合成及部分天然产物全合成时的关键步骤[3], [8]。

基本文献
- [1] A. Yasuda, S. Tanaka, K. Oshima, H. Yamamoto, H. Nozaki, J. Am. Chem. Soc. 1974, 96, 6513. doi: 10.1021/ja00827a044.
- [2] A. Yasuda, H. Yamamoto, H. Nozaki, Bull. Chem. Soc. Jpn. 1979, 52, 1705. doi: 10.1246/bcsj.52.1705.
- [3] H. Yamamoto, Tetrahedron 2007, 63, 8377. doi: 10.1016/j.tet.2007.05.128.
- [4] P. Thummel, B. Rickborn, J. Org. Chem. 1971, 36, 1365. doi: 10.1021/jo00809a011.
- [5] J. K. Crandall, H. Chang, J. Org. Chem. 1967, 32, 435. doi: 10.1021/jo01288a038.
- [6] C. Cope, A. Trumbull, A. PatriciaTrumbull, R. Elmer J. Am. Chem. Soc. 1958, 80, 2844-2849. doi: 10.1021/ja01544a063.
- [7] R. W. Thies, M. Gasic, D. Whalen, J. D. Grutzner, M. Sakai, B. Johnson, S. Winstein, J. Am. Chem. Soc. 1972, 94, 2262. doi: 10.1021/ja00762a015.
- [8] Y. Kitagawa, A. Itoh, S. Hashimoto, H. Yamamoto, H. Nozaki, J. Am. Chem. Soc. 1977, 99, 3864. doi: 10.1021/ja00453a069.
反应机理

参考文献
- [1] S. Bertillson, M. Sodergren, P. Andersson, J. Org. Chem. 2002, 67, 1567. doi: 10.1021/jo010934l.
- [2] C. Cope, H. Lee, E. Petree, J. Am. Chem. Soc. 1958, 80, 2849. doi: 10.1021/ja01544a064.
- [3] B. M. Trost, M. J. Bogdanowicz, J. Am. Chem. Soc. 1973, 95, 5311. doi: 10.1021/ja00797a036.
- [4] A. C. Cope, J. K. Heeren, J. Am. Chem. Soc. 1965, 87, 3125. doi: 10.1021/ja01092a021.
- [5] B. Rickborn, R. P. Thummel, J. Org. Chem. 1969, 34, 3583. doi: 10.1021/jo01263a078.
- [6] R. P. Thummel, B. Rickborn, J. Am. Chem. Soc. 1970, 92, 2064. doi: 10.1021/ja00710a045.
- [7] R. P. Thummel, B. Rickborn, J. Org. Chem. 1971, 36, 1365. doi: 10.1021/jo00809a011.
- [8] C. L. Kissel, B. Rickborn, J. Org. Chem. 1972, 37, 2060. doi: 10.1021/jo00978a002.
反应实例
链状烯丙醇的合成[1]-[2]

环烯丙醇的合成[1]

实验步骤
DATMP的原位生成:0 oC下,将二乙基氯化铝 (1 eq.)的苯溶液滴入到2,2,6,6-四甲基哌啶基锂 (1 eq.)的苯溶液中。将产生的浆状混合物搅拌30 min后,立即用于后续的Nozaki-Yamamoto消除过程。

Nozaki-Yamamoto消除过程:0 oC下,将原位生成的DATMP的苯溶液(4 eq., 0.4 M)滴加至环氧化物的苯溶液中(1 eq., 底物浓度为0.33 M),超过五分钟后,滴加结束。维持0 oC,将上述反应混合物继续搅拌,直至反应结束。反应结束后,加入冰冷却的1M HCl 淬灭反应。淬灭结束后,分出有机相,水相继续用乙醚进行萃取。将合并的有机相依次用饱和食盐水洗涤、无水硫酸钠干燥后,减压除去溶剂。粗产物采用制备TLC (乙醚/正己烷1:2v/v 作为展开剂)进行分离,获得相应目标产物。
参考文献
- [1] A. Yasuda, S. Tanaka, K. Oshima, H. Yamamoto, H. Nozaki, J. Am. Chem. Soc. 1974, 96, 6513. doi: 10.1021/ja00827a044.
- [2] A. Yasuda, H. Yamamoto, H. Nozaki, Bull. Chem. Soc. Jpn. 1979, 52, 1705. doi: 10.1246/bcsj.52.1705.
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!
本文作者:孙苏赟
继续上次的脱水反应。
(1) Burgess试剂
Burgess试剂是一种非常高效的商业化的试剂,可以和2o和3o醇发生反应进行脱水,且反应条件温和高效,但试剂储存需要在严格除水的条件下。大多数情况下,反应中的消除过程是syn-消除,但是也有不多的过anti-消除的例子的报道。对于3o醇来说,如果使用酸性条件下的阳离子机理的脱水反应,很可能导致发生碳正离子重排,但是用Burgess试剂就可以避免这个问题。
反应机理[1]:
这里是几个应用:
- Burgess试剂作用下的脱水不会导致两环中任一环发生缩环,且反应是倾向于生成多取代的双键化和物[2]
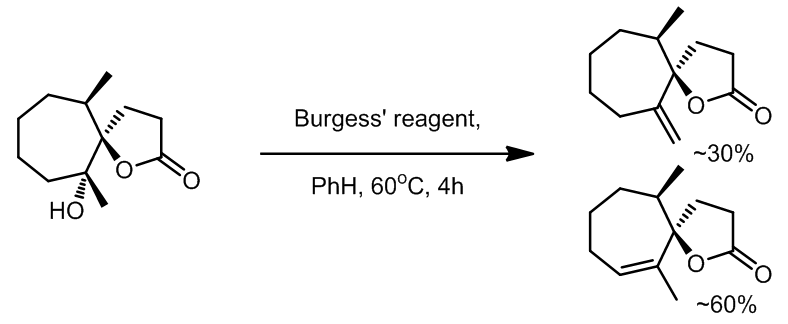
- 一个由于构型而导致发生anti-消除的例子 [3]
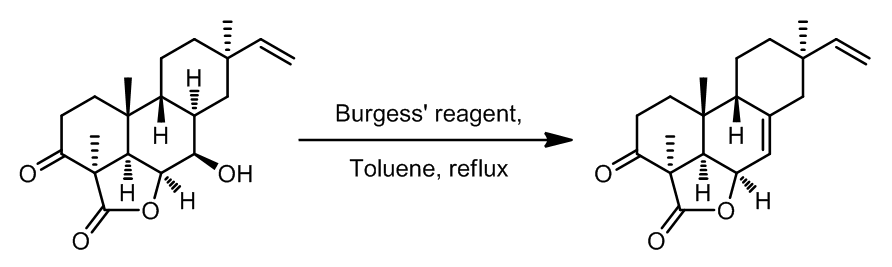
- [4]
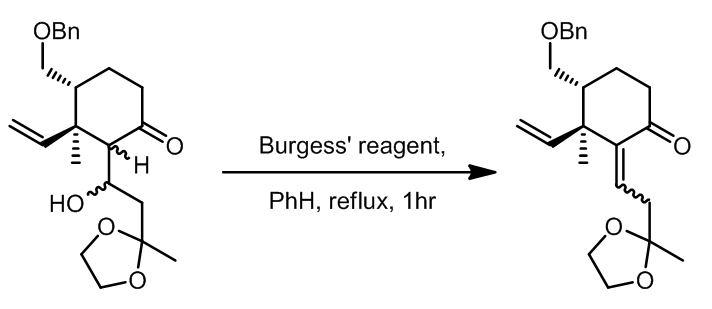
- 在Taxol的首次全合成中,Halton课题组在C环修饰的过程中使用到了Burgess试剂,反应得到的双键便于接下来构造Taxol中的D环结构。[5]
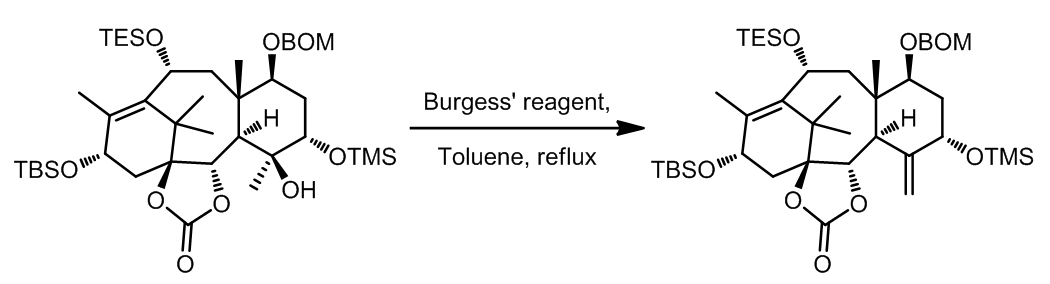




(2) Chugaev消除
利用醇对应的黄原酸酯的高温裂解过程得到双键的过程即为Chugaev消除反应,也成黄原酸酯裂解反应。底物醇的选择范围也较广,通常来讲1o醇的黄原酸酯对热较稳定,需要大于200oC的高温才可发生裂解,而2o和3o醇的黄原酸酯远次之。
反应机理:
从反应上来看,黄原酸酯的裂解和醋酸酯的裂解是类似的,但前者反应所需的温度较低,且反应过程的双键异构化几率较小,因此这个反应对于不太稳定的醇化合物通过非碳正离子过程转化成双键是非常有用的。但是对于双键位置的区域选择性,Chugaev反应显示出结果的选择性却不是太好。
几个例子:
- [6]
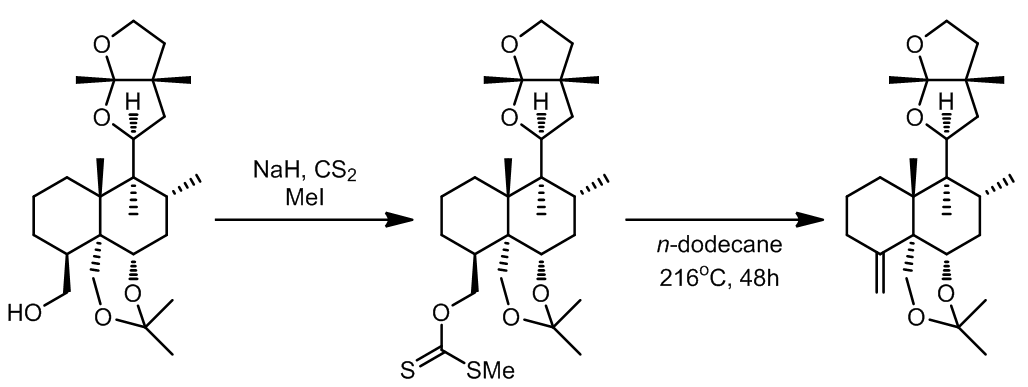

除此之外,Groot课题组尝试了许多种方法均未能将底物转化为环外双键,而黄原酸酯裂解的方法可以一步错的收率得到目标产物。
b. [7] c. 一种黄原酸酯的变体化合物 [8]
c. 一种黄原酸酯的变体化合物 [8]
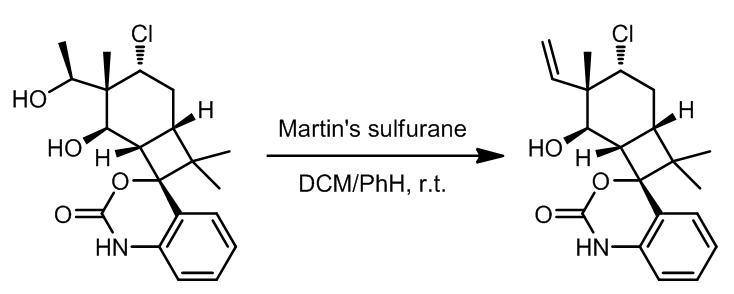
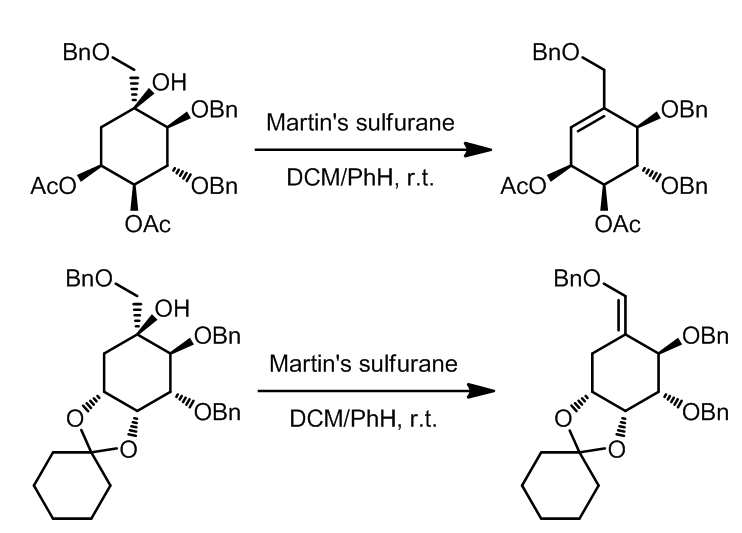
(3) Martin试剂
Martin试剂也称Martin Sulfurate,是一种已经商品化的试剂,它可以高效温和的将醇转化成对应的烯烃,即使在室温下也可快速发生反应。
反应机理:
Martin试剂也可与二醇反应得到环醚化合物。同时,此类试剂还有一种替代:
这里是几个实例:
- 由于两个羟基的位阻不同,因此反应活性上具有差别,位阻小者优先参与反应[9]
- [10]


(4) Lebedev Process
Lebedev反应是在钽试剂的作用下将乙醇发生脱水偶联得二烯的过程。早在二战时期,前苏联和纳粹德国军利用Lebedev反应来制造丁二烯作为生产工业橡胶的原料。反应机理大致如下:
由于高温的反应条件,事实上这个过程在有机合成中的利用价值不是太大。
REFERENCES
- J. Am. Chem. Soc.1968, 90, 17, 4744-4745, doi: 10.1021/ja01019a052
- Tetrahedron Lett., 1987, 43, 5287, doi: 1016/S0040-4020(01)87705-X
- J. Org. Chem., 2002, 67, 15, 5269-5278, doi: 10.1021/jo025873l
- J. Org. Chem., 1995, 60, 24, 7837-7848, doi: 10.1021/jo00129a025
- J. Am. Chem. Soc., 1994, 11, 64, 1597-1598, doi: 10.1021/ja00083a066
- J. Org. Chem., 1999, 64, 25, 9178-9188, doi: 10.1021/jo991151r
- Org. Lett., 2008, 10, 7, 1437-1440, doi: 10.1021/ol800259s
- J. Org. Chem., 1988, 5, 33, 477-481, doi: 10.1021/jo00238a003
- J. Am. Chem. Soc., 2006, 12, 85, 1448-1449, doi: 10.1021/ja057640s
- J. Org. Chem., 2001, 66, 21, 7184-7190, doi: 10.1021/jo010202t
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!
本文作者:孙苏赟
卤原子和磺酸酯在一定的条件下都是不错的离去基团,除了取代反应,他们也可以作为消除反应的底物。
- 脂肪卤代烃和磺酸酯
(1) 碱作用下的消除反应 (Zaitsev消除)
对于消除反应来说,无论反应是通过E1,E2机理,还是E1cb机理,都会比预期的更难发生,并且会产生多种消除异构体,包括区域异构体和Z/E-异构体。通常来说反应的产物是取代较多的较稳定的双键(Zaitsev规则),此外消除也会和亲核取代有竞争反应。
消除反应一般有一下几个影响因素:
- 反应底物:
a.1o和2o卤代烃和磺酸酯:E2消除,立体特异性的反式消除过程;
b. 3o,苯甲醇类:E1消除
c. α-卤代羰基化合物:E1cb消除 - 离去基团:羟基的离去性质较差,通常对于E2和E1cb反应会需要活化转化成其他的基团,酸性条件下可以进行E1消除。
- 碱:
大体积的碱有利于消除,减少取代;
常用的有:

几个例子:
- 碘代烃的消除[1]
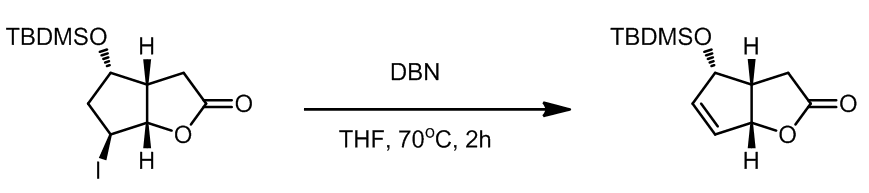 这一步是Corey课题组的prostaglandin的全合成的中的重要步骤,消除只会产生一种异构体,选择性非常好。
这一步是Corey课题组的prostaglandin的全合成的中的重要步骤,消除只会产生一种异构体,选择性非常好。 - 溴代烃的消除[2]
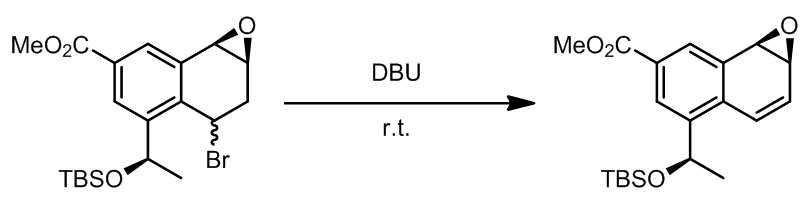


反应中,环氧化物并没有发生开环反应。
(2) 还原消除反应
这个反应中,首先是过渡金属和卤代烃发生氧化加成,之后可以非常顺利的进行β-H消除。但其实这样的β-H消除在交叉偶联反应中是不希望发生的副反应,但是却可以温和地用作消除卤素得到双键。
这个反应的应用不是很多,这里一个例子:[3]
- 几种脱水反应
(1) Grieco消除反应
Grieco消除反应中使用到了硒试剂作为氧化剂,反应中发生的是热力学消除,反应对于1o醇效果非常好。反应中试剂活化的过程和Mitsunobu反应类似,此外第二步的氧化过程中氧化剂的选择范围很广,常见的有H2O2,m-CPBA,等。
反应机理:
例如:
- [4]
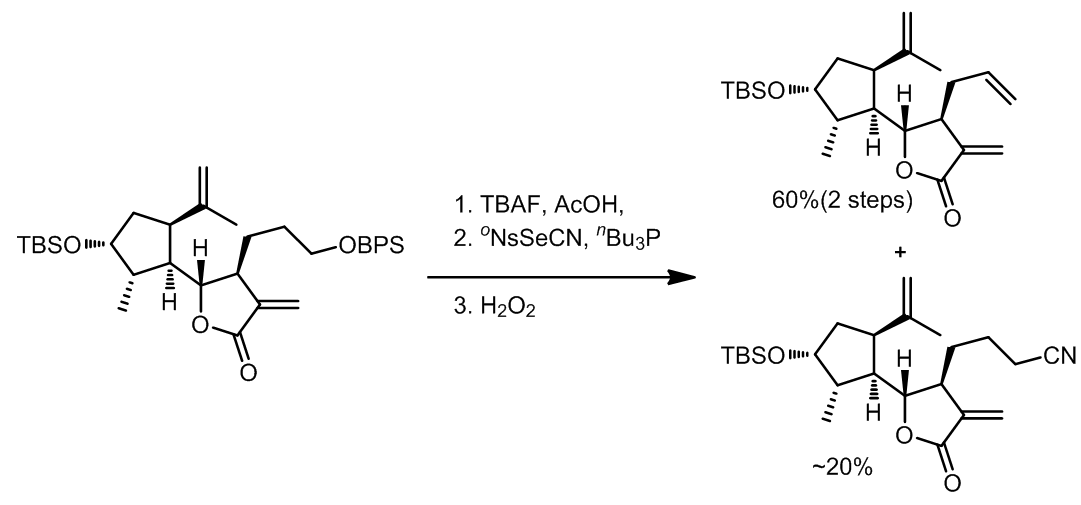

由于反应过程中会产生氰负离子,因此有一定的几率会产生氰代的副产物。
b. [5]
c. [6, 7]
(2) Bogert-Cook Synthesis[8]
这个反应是由Bogert课题组和Cook课题组几乎同时在1993年报道的,反应过程是Grignard试剂和酮反应生成得到了一个并三环化合物。这是在质子酸条件下羟基脱水经历碳正离子的一个很好的例子,当反应中间体和一个温和的脱水试剂接触时,反应的后一阶段脱水得到双键,并且成环,但是成环过程中具体的位置是和反应底物性质有关。
REFERENCES
- J. Am. Chem. Soc., 1973, 95, 20, 6832-6833, doi: 10.1021/ja00801a053
- J. Am. Chem. Soc., 2005, 127, 31, 11159-11175, doi: 10.1021/ja0529337
- J. Am. Chem. Soc., 2012, 134, 34, 14232-14237, doi: 10.1021/ja306323x
- J. Am. Chem. Soc., 2010, 13, 25, 1488-1489, 10.1021/ja9104478
- Tetrahedron Lett. 2004, 45, 3783. doi :1016/j.tetlet.2004.03.085
- J. Am. Chem. Soc. 1981, 103, 3213, doi: 10.1021/ja00401a050
- J. Am. Chem. Soc. 1981, 103, 3215, doi: 10.1021/ja00401a051
- Science, 1933, 77, 289, doi: 10.1126/science.77.1994.289
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!
概要
消除反应(Elimination Reaction)通常定义为从一个分子中失去部分原子或者官能团,形成多键的反应。 离去基团链接的碳(α碳)的邻位(β位)的氢被脱氢后一起离去的反应被称为β消除反応,拔氢的氢与离去基在同一个碳上的消除反应被称为α消除反应。通过α消除反应可以生成卡宾。
基本文献
反応机理
<β消除的分类>
脂肪族碳氢的β消除反应的机理,根据其分类差别还是很大的。
①单分子消除反应(E1反应)
・与SN1反应一样,首先离去基团解离后形成碳正离子中间体,然后碱再拔去β氢的分两步走的机理。 ・ 一般情况下离去基团的解离过程是该反应的限速阶段。反应速度只与底物的浓度有关(与碱的浓度无关)。 ・根据反应条件可能竞争性的伴有SN1反应或者碳正离子的重排反应(Wagner-Meerwein重排)的副产物。
・在碳正离子中间体十分稳定的情况下,容易发生异构化,因此该反应不利于立体选择性的控制。 
②二分子消除反应(E2反应)
・碱的拔氢与离去基的解离同时进行(协同的)的情况,被称为E2反应。 ・反应速度与底物跟碱的浓度相互依存。 ・α碳与β碳之间可以自由旋转的时候,离去基L与β在同一平面并且是anti的立体位置(antiperiplanar)情况下进行消除反应(anti消除)。这样由于轨道的重叠是最大的所以有利于π的形成。反应具有立体特异性。 ・在使用小位阻的碱拔氢的条件下,经常会伴随有竞争性的亲核取代(SN2反応)副反应。

・当底物的构型自由度比较低,比较难翻转形成anti-periplanar构型或者比较难进行分子间拔氢的情况下,也有可能进行synperiplanar消除反应(syn消除)。而在消除反应发生的时候,底物的轨道重叠也是尽可能达到最大化来辅助消除反应的进行,下图中给出了一个实例。

③共轭碱单分子消除反应(E1cB反应)
・区别于上述两种主要的消除反应、该机理首先底物上的β氢被碱拔去后生成碳副离子中间体,然后离去基团作为阴离子离去并且形成双键的机理(具体参照下图)。这种顺序的消除反应被称为E1cB反应,一般情况下离去基L的解离是该反应的限速阶段。
 <消除的位置选择性问题>
<消除的位置选择性问题>
热力学控制的立体选择性遵循Zaitsev法则,也就是生成取代数多的烯烃产物(参考:Angew. Chem. Int. Ed. 2009, 48, 5724.)。该法则也被称为Markovnikov法则的你法则。另外取代基多的烯烃热力学稳定的理由,可以通过超共轭机理进行解释。简单的说就是烯烃的π*轨道被越多的取代基的σ轨道的电子影响(共有倾向)的话,越稳定。

但是也有例外,在离去基是高位阻的四级胺基正离子的情况,或者使用大位阻的t-BuO–等碱的情况,生成的产物就不遵循Zaitsev法则,一般生成逆法则的产物(Hofmann消除)。
反应实例
实验步骤
实验技巧
参考文献
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载
概要
β-碳上有氢的氧化胺在加热的条件下发生syn消除,生成羟胺和烯烃。消除的方向遵循Hofmann法则。
基本文献
- Cope, A. C., Foster, T. T., Towle, P. H. J. Am. Chem. Soc. 1949, 71, 3929. DOI:10.1021/ja01180a014
- Cope, A. C.; LeBel, N. A. J. Am. Chem. Soc. 1960, 82, 4656. DOI: 10.1021/ja01502a052
- Cope, A. C.; Ciganek, E.; Howell, C. F.; Schweizer, E. E. J. Am. Chem. Soc. 1960, 82, 4663 doi:10.1021/ja01502a053
- Cope, A. C., Trumbull, E. R. Org. React. 1960, 11, 317.
- DePuy, C. H.; King, R. W. Chem. Rev. 1960, 60, 431. DOI: 10.1021/cr60207a001
反应机理

反应实例
实验步骤
实验技巧
参考文献
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!
概要
醇转化成Xanthate后,通过加热引起的syn-消除反应得到对应烯烃的手法。该反应也被称作黄原酸酯热分解反应(Xanthate Ester Pyrolysis)。
基本文献
- Chugaev, L. Ber. 1898, 31, 1775.
- Chugaev, L. Ber. 1899, 32, 3332.
- Chugaev, L. Ber. 1900, 33, 3118. DOI:10.1002/cber.19000330363
- DePuy, C. H.; King, R. W. Chem. Rev. 1960, 60, 431. DOI: 10.1021/cr60207a001
- Nace, H. R. Org. React. 1962, 12, 57.
开发历史
1898由俄罗斯化学家Chugaev开发报道。

反应机理

反应实例
实验步骤
实验技巧
参考文献
- Nakagawa, H.; Sugahara, T.; Ogasawara, K. Org. Lett. 2000, 2, 3181. DOI:10.1021/ol006377r
- Meulemans, T. M.; Stork, G. A.; Macaev, F. Z.; Jansen, B; J. M.; de Groot A. J. Org. Chem. 1999, 64, 9178. DOI: 10.1021/jo991151r
- Hagiwara, H.; Kobayashi, K.; Miya, S.; Hoshi, T.; Suzuki, T.; Ando, M. Org. Lett. 2001, 3, 251. DOI:10.1021/ol006893h
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!
- 概要
螯变反应是周环反应的一种。在同一原子上形成两个结合键的成环反应,或者它的逆反应,都被称为螯变反应。螯变反应形式上是(m+1)环加成反应,反应产物大多为五元环。
最有代表性的代表的螯变反应有1,3-丁二烯与二氧化硫的加成反应、环戊二烯酮的脱羰基反应以及卡宾跟烯烃的环丙烷化反应等。
- 基本文献
- Woodward, R.B.; Hoffman, R. Angew. Chem. Int. Ed. Engl. 1969, 8, 781. doi:10.1002/anie.196907811
- 反应机理
- 反应实例
通过螯变反应生成二烯的实例[1]

通过Ramberg-Backlund重排的脱SO2反应也被视作螯变反应。

分子内[4+1]加成反应实例[2]

- 实验步骤
- 实验技巧
- 参考文献
[2] (a) Spino, C.; Rezaei, H.; Dupont-Gaudet, K.; Belanger, F. J. Am. Chem. Soc. 2004, 126, 9926. DOI:10.1021/ja046344x (b) Boisvert , L.; Beaumier, F.; Spino, C. Org. Lett. 2007, 9, 5361. DOI: 10.1021/ol702172t
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!
醇→烯
- 概要
邻二醇与硫羰基二唑咪反应生成环状硫代碳酸酯,再用烷基亚磷酸酯处理发生顺式还原热消除得到相应的烯烃的方法。
把烯烃转换成二醇的方法很多,但是其逆反应却比较少,实用例子也很匮乏。对于羰基比较多的糖合成化学来说,该方法特别有用。
- 基本文献
・Corey, E. J.; Winter, R. A. E. J. Am. Chem. Soc. 1963, 85, 2677. doi:10.1021/ja00900a043
・Corey, E. J.; Hopkiss, P. B. Tetrahedron Lett. 1982, 23, 1797.
・Block ,E. Org. React. 1984, 30, 457.
- 反应机理
该反应被认为在过程中形成卡宾中间体。(参考: J. Org. Chem. 1970, 35, 3558)

- 反应实例
例[1]

使用二甲基甲酰胺二甲缩醛为反应物的类似变换手法。[2]

- 实验步骤
- 实验技巧
- 参考文献
[2] Hanessian, S.; Bargiotti, A.; La Rue, M. Tetrahedron Lett. 1978, 737.
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载
- 概要
利用Methyl N-(triethylammoniumsulfonyl)carbamate (Burgess试剂)试剂,与经典的脱水反应(酸或碱催化下加热)不同的是,可以在低温以及中性的温和条件下使底物进行脱水反应。但是无法用于一级醇的脱水反应。
- 基本文献
・Atkins, G. M.; Burgess, E. M. J. Am. Chem. Soc. 1968, 90, 4744. doi:10.1021/ja01019a052.
・Burgess, E. M.; Penton, H. R., Jr.; Taylor, A. E. J. Am. Chem. Soc. 1970, 92, 5224. DOI: 10.1021/ja00720a041
・Burgess, E. M.; Penton, H. R.; Taylor, E. A. J. Org. Chem. 1973, 38, 26. DOI: 10.1021/jo00941a006
・Burgess, E. M.; Penton, H. R. Jr; Taylor, E. A.; Williams, W. M. Org. Synth. 1977, 56, 40. [PDF]
・Khapli, S.; Dey, S.; Mal, D. J. Indian Inst. Sci. 2001, 81, 461. [PDF]
- 反应机理
分子内syn消除(Ei)机理。

- 反应实例
该试剂不仅能用于烯的合成,也能用于腈,异腈以及氧化腈的合成。此外它还被用于合成杂环恶唑啉和噻唑啉。[1]

Fredericamycinの合成[2]

Taxolの合成[3]

Penitrem Dの合成[4]

- 实验步骤
Burgess试剂的制备方法[5]

- 实验技巧
※由于该试剂的吸湿性比较高,所以容易失活。所以使用与保存方面需要注意。
- 参考文献
[2] (a) Kita, Y. et al. Angew. Chem. Int. Ed. 1999, 38, 683.[abstract] (b) Kita, Y. et al. J. Am. Chem. Soc. 2001, 123, 3214. DOI: 10.1021/ja0035699
[3] (a) Holton, R. A. et al. J. Am. Chem. Soc. 1994, 116, 1597. doi: 10.1021/ja00083a066 (b) Holton, R. A. et al. J. Am. Chem. Soc. 1994, 116, 1599. doi:10.1021/ja00083a067
[4] (a) Smith, A. B., III et al. J. Org . Chem. 1995, 60, 7837. DOI: 10.1021/jo00129a025 (b) Smith, A. B., III et al. J. Am. Chem. Soc. 2003, 125, 8228. DOI: 10.1021/ja034842k
[5] (a) Burgess, E. M.; Penton, H. R.; Taylor, E. A. J. Org. Chem. 1973, 38, 26. DOI: 10.1021/jo00941a006 (b) Burgess, E. M.; Penton, H. R. Jr; Taylor, E. A.; Williams, W. M. Org. Synth. 1977, 56, 40. [PDF]
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载