概要
Naito吲哚合成 (Naito indole synthesis)是Nʹ-芳基-N-三氟乙酰基烯肼 (Nʹ-aryl-N-trifluoroacetyl enehydrazine)在加热条件下依次通过环化以及热消除反应过程,形成吲哚类化合物的反应。该反应由日本Kobe药科大学 (神戸薬科大学, Kobe Phamaxutical University) 的Naito (内藤 猛章,Naito Takeaki )研究室在1999年首次报道[1]。

2001年,Naito进一步研究发现,通过各类酮与肼之间的一锅反应,同样能够顺利完成吲哚类化合物的合成[2]。反应过程中无需中间体的分离,操作简便,具有良好的收率与pot-economy[2]。

2002年,Naito进一步将底物范围扩展至N-三氟乙酰基-N’-(3-甲氧羰基苯基)烯肼,并以中等程度的收率获得相应2,4-二取代吲哚产物[3]。

2003年,Naito继续将上述反应条件扩展至苯并呋喃环的构建,并获得良好的反应收率[4]。

Naito吲哚合成方法学具有条件温和,目标产物产率优良等优点。为吲哚环结构的构建以及部分生物活性分子的构建开辟了一种更为简便的方法[5]-[6]。
基本文献
- [1] O. Miyata, Y. Kimura, K. Muroya, H. Hiramatsu, T. Naito, Tetrahedron Lett. 1999, 40, 3601. doi: 10.1016/S0040-4039(99)00583-3.
- [2] O. Miyata, Y. Kimura, T. Naito, Synthesis, 2001, 1635. doi: 10.1055/s-2001-16756.
- [3] O. Miyata, N. Takeda, T. Naito, Heterocycles 2002, 57, 1101. doi: 10.3987/COM-02-9476.
- [4] O. Miyata, N. Takeda, Y. Morikami, T. Naito, Org. Biomol. Chem. 2003, 1, 254. doi: 10.1039/B210059B.
- [5] O. Miyata, N. Takeda, Y. Kimura, Y. Takemoto, N. Tohnai, M. Miyata, T. Naito, Tetrahedron 2006, 62, 3629. doi: 10.1016/j.tet.2006.01.087.
- [6] O. Miyata, N. Takeda, T. Naito, Org. Lett. 2004, 6, 1761. doi: 10.1021/ol049564o.
反应机理

参考文献
- [1] O. Miyata, Y. Kimura, T. Naito, Chem. Commun. 1999, 2429. doi: 10.1039/A907539I.
- [2] O. Miyata, N. Takeda, Y. Kimura, Y. Takemoto, N. Tohnai, M. Miyata, T. Naito, Tetrahedron 2006, 62, 3629. doi: 10.1016/j.tet.2006.01.087.
- [3] O. Miyata, N. Takeda, Y. Kimura, Y. Takemoto, N. Tohnai, M. Miyata, T. Naito, Tetrahedron 2006, 62, 3629. doi: 10.1016/j.tet.2006.01.087.
反应实例
吲哚的合成[1]

苯并呋喃的合成[2]

eupomatenoid 6的合成[3]

实验步骤
将Nʹ-芳基-N-三氟乙酰基烯肼溶于适量甲苯 (维持底物浓度为0.008 M)中。将上述均相混合物加热回流,并通过TLC监控,直至反应结束。反应结束后,将上述混合物减压浓缩。残余物通过MPCC (medium-pressure column chromatography)进行分离纯化 (正己烷/乙酸乙酯 5 : 1 v/v 作为洗脱剂), 获得相应吲哚产物。
参考文献
- [1] O. Miyata, Y. Kimura, K. Muroya, H. Hiramatsu, T. Naito, Tetrahedron Lett. 1999, 40, 3601. doi: 10.1016/S0040-4039(99)00583-3.
- [2] O. Miyata, N. Takeda, Y. Morikami, T. Naito, Org. Biomol. Chem. 2003, 1, 254. doi: 10.1039/B210059B.
- [3] O. Miyata, N. Takeda, T. Naito, Org. Lett. 2004, 6, 1761. doi: 10.1021/ol049564o.
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!
概要
Kihara吲哚合成 (Kihara indole synthesis)是通过强碱(正丁基锂)条件下原位生成的芳基锂试剂进行的分子内Barbier反应,进而完成的1-甲基-3-芳基吲哚与1-甲基-3-烷基吲哚及1-甲基-3-烷基-3-羟基吲哚的合成[1]。该反应由日本Tokushima大学药学部的Kihara (木原 勝, 徳島大学薬学部, Kihara Masaru, Faculty of Pharmaceutical Sciences, The University of Tokushima) 在1995年首次报道。该方法学原料易得,产率中等,为1-甲基-3-芳基吲哚与1-甲基-3-烷基吲哚及1-甲基-3-烷基-3-羟基吲哚的合成开辟了新的途径。

基本文献
- [1] M. Kihara, Y. Iwai, Y. Nagao, Heterocycles, 1995, 41, 2279. doi 10.3987/COM-95-7177.
反应机理

反应实例
1-甲基-3-芳基吲哚的合成[1]

1-甲基-3-烷基吲哚的合成[1]

1-甲基-3-烷基-3-羟基吲哚的合成[1]

实验步骤
1. Kihara吲哚合成
在-78 oC,氮气气氛下,将正丁基锂 (l.6 M 正己烷溶液,1.2 eq.)缓慢滴加至N-(2-碘苯基)-N-甲氨基甲基酮 (1 eq.)的无水四氢呋喃溶液 (底物浓度为0.11 M)中。维持-78 oC,将上述反应混合物搅拌15 min。15 min后,加入水淬灭反应。淬灭完成后,再加入乙醚进行萃取。将合并的有机相通过无水硫酸镁干燥后,减压除去溶剂。粗产物首先通过硅胶快速柱色谱 (DCM作为洗脱剂)分离出相应1-甲基-3-烷基-3-羟基吲哚组分,随后将该组分继续通过硅胶快速柱色谱 (DCM/正己烷 1:1v/v 作为洗脱剂)进行纯化,获得相应吲哚产物。
2. 在HCl/Et2O条件下的脱水
向1-甲基-3-烷基-3-羟基吲哚或1-甲基-3-苯基-3-羟基吲哚的乙醚溶液中加入适量5% HCl溶液。再将上述混合物在室温下搅拌10 min。之后,加入适量10% KOH进行中和,随后,加入DCM进行萃取。将合并的有机相加水洗涤,再通过无水硫酸镁进行干燥。减压除去溶剂后,获得相应吲哚产物。
参考文献
- [1] M. Kihara, Y. Iwai, Y. Nagao, Heterocycles, 1995, 41, 2279. doi 10.3987/COM-95-7177.
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!
概要
Arcadi-Cacchi反应 (Arcadi-Cacchi reaction)又称为Cacchi反应 (Cacchi reaction)或Cacchi吲哚合成 (Cacchi Indole Synthesis),是在钯催化剂存在下,邻炔基三氟乙酰苯胺与乙烯基三氟甲磺酸酯或芳卤反应,生成2,3-二取代吲哚的反应。该反应由意大利Sapienza Rome大学药物化学与技术系 (Department of Drug Chemistry and Technologies, Sapienza University of Rome)的S. Cacchi与A. Arcadi研究组在1992年首次报道[1]。

1997年,J. W. Ellingboe等将该反应成功应用于吲哚组合化合物库的固相合成[2]。

之后,S. Cacchi采用Pd2(dba)3催化剂,使反应温度进一步降低,同时,成功完成3-芳基吲哚骨架的构建[3]。

1998年,S. Cacchi通过该反应,顺利实现3-烯丙基吲哚的区域选择性合成[4]。

2000年,S. Cacchi采用Pd2(dba)3与三(2,4,6-三甲氧基苯基)膦配体,进一步将底物范围拓展至碘乙酸乙酯及苄基溴,并为吲哚羧酸酯及2-取代-3-苄基吲哚的合成提供了一种更为简便的方法[5]。

2003年,S. Cacchi将反应底物扩展到杂芳卤,同时,研究表明,该反应具有优良的官能团兼容性[6]。

2005年,S. Cacchi继续将反应底物扩展至1-炔基卤,以良好的产率成功获得2-取代3-炔基吲哚[7]。

2010年,S. Cacchi发现通过芳香重氮盐与邻炔基三氟乙酰苯胺之间的反应,同样可获得2,3-二取代吲哚,并且,反应条件较为温和,具有更为优良的官能团兼容性[8]。

2015年,Zhu与Luo等通过钯催化剂与邻炔基三氟乙酰苯胺的钯氨基化(aminopalladation)引发的carbene插入过程,成功实现了2-(1H-吲哚-3-基)乙酸酯的合成[9]。

Arcadi-Cacchi反应条件温和,具有优良的产率与官能团兼容性及广泛的底物适用性。目前,该反应已成为构建吲哚类化合物的一种十分简便快捷的方法[10]-[13]。
基本文献
- [1] A. Arcadi, S. Cacchi, F. Marinelli, Tetrahedron Lett. 1992, 33, 3915. doi: 10.1016/S0040-4039(00)74818-0.
- [2] M. D. Collini, J. W. Ellingboe, Tetrahedron Lett. 1997, 38, 7963. doi: 10.1016/S0040-4039(97)10140-X.
- [3] S. Cacchi, G. Fabrizi, F. Marinelli, L. Moro, P. Pace, Synlett 1997, 1363. doi: 10.1055/s-1997-1039.
- [4] S. Cacchi, G. Fabrizi, P. Pace, J. Org. Chem. 1998, 63, 1001. doi: 10.1021/jo971237p.
- [5] A. Arcadi, S. Cacchi, G. Fabrizi, F. Marinelli, Synlett, 2000, 394. doi: 10.1055/s-2000-6529.
- [6] S. Cacchi, G. Fabrizi, D. Lamba, F. Marinelli, L. M. Parisi, Synthesis 2003, 728. doi: 10.1055/s-2003-38079.
- [7] A. Arcadi, S. Cacchi, G. Fabrizi, M. Giancarlo, P. Fabio, M. Luca, J. Org. Chem. 2005, 70, 6213. doi: 10.1021/jo050517z.
- [8] S. Cacchi, G. Fabrizi, A. Goggiamani, A. Perboni, A. Sferrazza, P. Stabile, Org. Lett. 2010, 12, 3279. doi: 10.1021/ol101321g.
- [9] Z. Hu, S. Luo, Q. Zhu, Adv. Synth. Catal. 2015, 357, 1060. doi: 10.1002/adsc.201400799.
- [10] G. Battistuzzi, S. Cacchi, G. Fabrizi, Eur. J. Org. Chem. 2002, 2671. doi: 10.1002/1099-0690(200208)2002:16<2671::AID-EJOC2671>3.0.CO;2-X.
- [11] S. Cacchi, G. Fabrizi, Parisi, L. M. Synthesis 2004, 1889. doi: 10.1055/s-2004-815993.
- [12] S. Cacchi, G. Fabrizi, Chem. Rev. 2011, 111, PR215. doi: 10.1021/cr100403z.
- [13] K. Krüger, A. Tillack, M. Beller, Adv. Synth. Catal. 2008, 350, 2153. doi: 10.1002/adsc.200800409.
反应机理

参考文献
- [1] S. Cacchi, Fabrizi, Chem. Rev. 2005, 105, 2873. doi: 10.1021/cr040639b.
- [2] S. Cacchi, V. Carnicelli, F. Marinelli, J. Organomet. Chem. 1994, 475, 289. doi: 10.1016/0022-328X(94)84034-2.
反应实例
吲哚并[2,3-a]咔唑的合成[1]

2,3-二芳基吲哚的合成[2]

3-杂芳基吲哚的合成[3]

实验步骤
向Carousel Tube反应器 (Radely Discovery Technology生产)中加入邻苯乙炔基三氟乙酰苯胺 (1 eq.)的乙腈溶液 (底物浓度为0.17 M)、1-溴苯乙炔 (1.2 eq.)、Pd(PPh3)4 (0.05 eq.)及Cs2CO3 (1.5 eq.)。 将上述反应混合物在60 °C下搅拌,直至反应结束。反应结束后,将上述混合物冷却至室温,加入乙酸乙酯稀释,再加入水进行洗涤, 最后,采用无水硫酸钠进行干燥。减压除去溶剂后,将粗产物采用硅胶柱色谱分离纯化 (正己烷/乙酸乙酯9:1 v/v)后,获得相应吲哚产物。
参考文献
- [1] M. G. Saulnier, D. B. Frennesson, M. S. Deshpande, D. M. Vyas, Tetrahedron Lett. 1995, 36, 7841. doi: 10.1016/0040-4039(95)01644-W.
- [2] S. Cacchi, G. Fabrizi, F. Marinelli, L. Moro, P. Pace, Synlett 1997, 1363. doi: 10.1055/s-1997-1039.
- [3] A. Arcadi, S. Cacchi, and F. Marinelli, Tetrahedron Lett. 1992, 33, 3915. doi: 10.1016/S0040-4039(00)74818-0.
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!
概要
Natsume 吲哚合成(Natsume indole synthesis)是通过采用Lewis酸 (如AlCl3及 BF3·Et2O)参与的吡咯环的亲电取代反应,随后,经官能团化过程而进行的吲哚环的合成。该反应由日本Research Foundation Itsuu Laboratory的夏目充隆(Natsume Mitsutaka)研究室1990年首次报道。目前,该反应已经广泛应用于各类烷基取代吲哚类化合物的合成及吲哚类生物碱的全合成。
基本文献
[1] H. Muratake, M. Natsume, Heterocycles 1990, 31, 683. doi: 10.3987/COM-90-5333. [2] K. Okabe, H. Muratake; M. Natsume, Tetrahedron 1990, 46, 5113. doi: 10.1016/S0040-4020(01)87818-2.反应机理

反应实例
4-烷基吲哚的合成[1]

吲哚类生物碱pendolmycin的全合成[2]

实验步骤
- Friedel-Crafts酰基化[3] [4]
25oC下,将BF3•OEt2(2.2 eq.)加入到乙酸酐(1.1 eq.)的DCE溶液(底物浓度为0.36 M)中。将上述反应混合物搅拌10 min后,加入吡咯(1eq.)的DCE溶液(底物浓度为1M),将上述混合物在该温度下搅拌,直至反应结束。反应结束后,加入冷水淬灭反应,并用DCM进行萃取。将合并的有机相减压除去溶剂后获得粗产物。将粗产物采用硅胶柱色谱(乙酸乙酯/甲苯 1:4 v/v作为洗脱剂)进行分离纯化,获得相应乙酰基化产物。最后,在正己烷溶剂中进行重结晶获得纯净的酰基化产物。
- Grignard 反应
-20°C,氩气气氛下,将2-(1,3-二噁烷-2-基)乙基溴化镁(1eq.)的THF (浓度为溶液加入到酰基吡咯(1eq.)的THF (底物浓度为0.09 M)溶液中。将上述反应混合物搅拌15 min后,加入sat. NH4Cl/H2O淬灭反应,淬灭结束后,再加入DCM进行萃取。将合并的有机相采用无水硫酸钠进行干燥后,减压除去溶剂。粗产物采用Prep-TLC(正己烷/EtOAc 5:2 v/v作为展开剂)分离纯化获得相应成环前体。
- 吲哚成环反应
将第二步反应产生的成环前体(1eq.)的6% H2SO4-异丙醇溶液(底物浓度为0.03 M)加热回流30min。反应结束后,将反应液进行冷却后加水稀释,再加入DCM进行萃取。将合并的有机相采用无水硫酸钠进行干燥后,减压除去溶剂。粗产物采用Prep-TLC(正己烷/EtOAc 9:l v/v作为展开剂)分离纯化获得最终目标产物。
实验技巧
参考文献
- [1] H. Muratake, M. Natsume, Heterocycles 1990, 31, 683. doi: 10.3987/COM-90-5333.
- [2] K. Okabe, H. Muratake; M. Natsume, Tetrahedron 1990, 46, 5113. doi: 10.1016/S0040-4020(01)87818-2.
- [3] J. Rokach, P. Hamel, M. Kakushima, G. M. Smith, Tetrahedron Lett. 1981, 22, 4901. doi: 10.1016/S0040-4039(01)92377-9.
- [4] M. Kakushima, P. Hamel, R. Frenette, I. Rokach, J. Org. Chem. 1983,48, 3214. doi: 10.1021/jo00167a014.
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!
概要
1974年,美国Idaho大学(University of Idaho)的Thyagarajan首次报道了通过芳基炔丙胺氮氧化物连续的[2,3]与[3,3]-σ-重排获得吲哚类化合物的反应,称为Thyagarajan吲哚合成(Thyagarajan indole synthesis)。随后,Majumdar将该反应扩展到具有两个吲哚结构单元的环双醚及二氢-1H-吡喃并[3,2-e] 吲哚-7-酮的合成。
基本文献
- [1] B. S. Thyagarajan, J. B. Hillard, K. V. Reddy, K. C. Majumdar, Tetrahedron Lett., 1974, 15, 1999. doi: 10.1016/S0040-4039(01)82615-0.
- [2] J. Hillard, K. V. Reddy, K. C. Majumdar, B. S. Thyagarajan, J.Heterocycl. Chem., 1974, 11, 369. doi: 10.1002/jhet.5570110319.
- [3] B. S. Thyagarajan, K. C. Majumdar, J. Heterocycl. Chem., 1975, 12, 43. doi: 10.1002/jhet.5570120108.
- [4] K. C. Majumdar, G. H. Jana, U. Das, Chem. Commun., 1996, 517.doi: 10.1039/CC9960000517.
- [5] K. C. Majumdar, G. H. Jana, U. Das, J. Chem. Soc., Perkin Trans. 1, 1997, 1229. doi: 10.1039/A606047A.
- [6] K. C. Majumdar, S. K. Ghosh, J. Chem. Soc., Perkin Trans. 1, 1994, 2889. doi: 10.1039/P19940002889.
反应机理



反应实例
吡咯香豆素的合成[1]

N-烷基-2-乙烯基吲哚的合成[2]

实验步骤
将MCPBA (1 eq.)的三氯甲烷或二氯甲烷溶液(0.06 M)缓慢加入至芳基炔丙胺(1 eq.)的三氯甲烷或二氯甲烷溶液(0.05 M)中。将上述反应混合物在22-23oC下搅拌,直至反应结束。反应结束后,将反应液用10%碳酸钾溶液及饱和食盐水洗涤,并采用无水硫酸钠干燥。减压除去溶剂后,将粗产物采用二氯甲烷-石油醚(30-60oC)进行重结晶,获得最终目标产物。
实验技巧
参考文献
- [1] K. C. Majumdar, S. K. Ghosh, J. Chem. Soc., Perkin Trans. 1, 1994, 2889. doi: 10.1039/P19940002889.
- [2] T. Balasubramanian, K. K. Balasubramanian, J. Chem. Soc., Chem. Commun., 1994, 1237. doi: 10.1039/C39940001237.
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!
概要
Iwao 吲哚合成(Iwao indole synthesis)涉及N-叔丁氧羰基苯胺的在叔丁基锂作用下的DoM反应(Directed ortho metalation)、采用1-三烷基硅基-1-苯亚磺酰基乙烯对的有机锂中间体的淬灭、热sila-Pummerer 重排获得2-thioindolines、采用m-CPBA对2-2-thioindolines的氧化生及亚砜的syn-消除等五个反应过程,最终获得相应吲哚化合物。无Boc保护基的苯胺化合物及N-特戊酰基苯胺(在N-叔丁氧羰基-3-苯胺的DoM反应难以进行时,可采用N-特戊酰基苯胺可作为其替代物)同样可以参与该反应过程。该反应由日本长崎大学(Nagasaki University)的岩尾正伦(Iwao Masatomo)研究组在1994年首次报道。目前,该反应主要用于吲哚类杂环化合物的合成。

基本文献
[1] M. Iwao, Heterocycles, 1994, 38, 45. doi: 10.3987/COM-93-6563.
反应机理

反应实例
吲哚及吲哚林类化合物的合成[1]

实验步骤
吲哚啉类化合物的合成[1] [2]:在-78oC,氮气气氛下,将2M 的叔丁基锂 (2.4 eq.)的戊烷溶液滴加至N-叔丁氧羰基苯胺 (l eq.) 的无水THF溶液(0.4 M)中。搅拌15 min后,将上述溶液升温至-20oC,继续搅拌3 h。再向上述溶液中加入1-三烷基硅基-1-苯亚磺酰基乙烯(1.5 eq.),并继续在-20oC下搅拌2 h。反应结束后,采用sat. NH4Cl溶液淬灭反应,后将反应液用乙酸乙酯进行萃取,再将萃取液回流1 h。减压除去溶剂后,将粗产物采用硅胶柱色谱(正己烷/乙酸乙酯 20:1 v/v 作为洗脱剂)分离纯化,获得相应吲哚啉类化合物。
吲哚啉类化合物的氧化[3]:将吲哚啉(l eq.)的二氯甲烷溶液(0.05 M)冷却至-78oC,后用注射器将间氯过苯甲酸(l eq.)的二氯甲烷溶液(0.15 M)滴加至上述均相溶液中(超过5 min后,滴加完毕)。滴加完毕后,开启搅拌,并用TLC监控,直至反应完成。反应结束后,将冷的反应液倒入装有乙醚及10%亚硫酸钠溶液的分液漏斗中,萃取分出有机相。再将有机相用饱和碳酸氢钠溶液洗涤、干燥,减压蒸馏除去溶剂后,将所得粗产物直接进行后续的消除反应。
吲哚啉类化合物的消除[3]:
将氧化反应所得粗产物(l eq.)溶于甲苯(0.13 M)中,将上述反应混合物加热至回流,并用TLC监控,直至反应完成。反应结束后,减压蒸馏除去溶剂。将所得粗产物采用硅胶柱色谱(三氯甲烷作为洗脱剂)分离纯化,获得最终目标产物。
参考文献
[1] M. Iwao, Heterocycles, 1994, 38, 45. doi: 10.3987/COM-93-6563. [2] J. M. Muchowski, M. C. Venuti, J. Org. Chem. 1980,45, 4801. doi: 10.1021/jo01311a059. [3] B. M. Trost, T. N. Salzmann, K. Hiroi, J. Ame. Chem. Soc., 1976, 98, 4887. doi: 10.1021/ja00432a034.实验安全须知
小编郑重提示:叔丁基锂接触空气时,极易发生自燃,实验操作时需要格外小心,严格避免与空气接触,最好在氩气手套箱内进行操作。
参考文献
[1] M. Iwao, Heterocycles, 1994, 38, 45. doi: 10.3987/COM-93-6563.
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!
本次的专访请到的是由化学空间小编推荐,来自静冈县立大学药学部,药化学教室教授——真锅敬先生。主要以促进有机反应催化相关的「催化剂」为研究课题,以开发实现较难分子之间转换的催化剂为目标。不仅如此,正如题目中所指,真锅教授的研究为实现安全 简便 短工程而开发催化反应为主,特别是最近开发的试剂均已在各试剂公司开始上市销售。
真锅教授究竟对化学这条研究道路心怀何志呢?让我们一起来走进真锅老师的化学世界吧。
Q. 您为什么要从事化学研究
不好意思地说,进入大学学习之前并没有觉得化学是多么有意思的科目。高中化学让我觉得略有些无趣。特别是有机化学部分几乎没有留下什么印象。但是,在作为大学本科生阶段时,有幸聆听了来自农学部的森谦治老师的有机化学课,从此对化学的想法发生了巨大转变。在那堂课上,森先生介绍了环己烷的椅式构型的立体书写方法,先生说到「这并不是化学,这简直就是制图」。只要注意到哪些结构是平行的,就可以得心应手地画出,我想先生是想传达出这样的窍门吧。但是,仅仅因为先生的一句话,让我产生了不单单止于窍门的思想碰撞。第一次感受到了有机化学的理论性、有了一种「啊!我懂了!」的感觉,第一次发现「有机化学很有意思!」,我想也是以此为开端选择了成为化学研究者这条路吧。

Q. 如果不从事化学,还有什么想做的?为什么?
虽说自知没有那个能力,不过曾经很想成为漫画家或小说家。很喜欢能创造出自己的作品。小学低年级的时候很想成为漫画家,在家里也经常画一些漫画。不久前回老家的时候还发现了小时候画的一些漫画。现在看上去画得很糟糕。中学时代很想成为小说家。确实,那时候筒井康隆先生,模仿着流行作家的写作方法写一些文字朋友们读后颇受刺激。那时候流行着星新一先生的短篇小说,自己也想这样的一些短篇的话自己也能写得出来吧,结果完全不行。
Q. 现在在做哪方面的研究?另外,准备在这块如何展开?
以「安全・简便・短工程」为关键,利用催化剂的有机合成反应的相关研究。主要围绕以下两个研究项目。
(1)利用一氧化碳的代替物进行催化反应的开发
一氧化碳(CO)是被较为广泛使用的C1原料,因其性质为具有毒性的气体在使用的时候需要格外注意。而与此同时,能方便使用以及在反应体系内生成CO的「CO代替化合物」引起了更多关注。在这一领域中的先驱研究有(T. Morimoto and K. Kakiuchi, Angew. Chem. Int. Ed. 2004, 43, 5580. DOI: 10.1002/anie.200301736 等)。我在任静冈县立大学此研究领域以来也参与了此研究领域中。通过多次试验,发现一些甲酸衍生物具有在弱碱性条件存在下可分解出CO。利用此类乙酸衍生物可代替CO,将安全简便地催化羧基化反应成为现实。到目前为止,这样的甲酸衍生物(苯甲酸类,三氯苯甲酸、甲酰基糖精)等均以上市销售。今后,我希望能为更多的使用者在各种反应中使用不断改良。此外,最近不仅仅是CO的替代物,我们也开始着手于利用其他类的毒性气体的替代物的催化反应研究。能为「安全・简便的催化反应」这一领域的发展作出一些贡献,研究室的工作人员以及学生们正在不懈努力中。

(2)实现位置选择性催化反应的开发
位置选择性的过渡金属触媒反应主要是从容易得到的原料开始,通过短工程合成化合物的一种有效方法。在这个研究过程,我们开发了新型磷配位子DHTP。研究过程中发现,DHTP与Pd组合后作为催化剂使用,可使二卤代化合物的偶联反应得到位置选择性反应结果。此外,我们还利用此催化剂开发了置换苯并呋喃或置换型吲哚的一锅合成法。
DHTP为由来的催化剂中,具有基质活性化部位和基质捕捉部位相匹配的bifunctional catalyst的机能。最近,DHTP也得到上市销售。今后,在扩展DHTP的应用范围的同时,可以通过活用此催化剂设计,开发能实现短工程有机合成的新规催化剂。

Q. 如果能和历史上的某一个人共进晚餐的话,您最希望是谁?为什么?
说不上是历史上的人物,我很想再见见我的指导教师古賀憲司先生。就如同常说的「子欲养而亲不待」,在学问研究上,古賀先生就犹如我的父亲一样。我想,曾在先生生前总是做些失礼的事情。如果是换成现在自己的学生,我想自己会很生气吧。很想见老师一面,向老师道歉,也感谢老师的培育之恩。

Q. 您最后一次在实验室做实验是什么时候?什么研究内容?
翻看了一下实验笔记是在2012年10月16日。到那之前还在做着一些实验。最后的实验应该是为发现研究课题,反应也是不成样子好像只看了看TLC就扔掉了。
Q.当您搁浅在了一个荒岛上、怎样的书与音乐是必须的?只能说一样。
应该是加缪的《西西弗斯的神话》吧。着实被这本书的最终章节感动了。书中告诉我们,希腊神话中的出场人物西西弗斯被被判永久的将一块大石头推上山顶,永不停息地重复重体力的惩罚。即便如此他依旧找到了活着的意义,甚至感到了幸福的价值。如果真是到了无人的沙漠的话,这本书能带给我这样的感觉。
[amazonjs asin=”B009IF6002″ locale=”CN” title=”西西弗斯的神话”]
// <![CDATA[
document.write("
“)
// ]]>
Q. 下一次您推荐我们采访谁。
有很多,必须要推荐的是京都大学的川端猛夫先生。作为化学研究者都非常尊敬的、也是我非常喜欢的一位教授
相关链接
- 静岡県立大学薬学部薬化学教室
- 位置選択的カップリング配位子Cy-DHTP・HBF4|和光純薬
- 新規一酸化炭素等価体を用いる実用的カルボニル化反応の開発 (PDFファイル)| 和光純薬Organic Square
真锅敬教授 简历

1965年生,1988年东京大学药学部毕业,其后升入同大学药学研究科理学研究科博士课程,1998年获博士(药学)学位。同年于东京大学药学部从事助手工作,2000年任讲师、助教授、2005年理化学研究所主干研究员。期间于1993年哥伦比亚大学担任化学科博士研究员。2009年后至今任静冈县立大学教授。
研究领域为有机化学,主要从事新规化学合成法的开发、催化反应、技能性物质合成等研究。
1998年获有机合成化学协会研究企划奖、2001年日本药学会奖励奖等
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载
- 概要
醛,酮和芳基联氨反应,生成的芳基腙在酸性催化剂下加热,经过西格玛迁移环化生成取代基吲哚的反应。
生物碱和药物合成中不可缺少的常用合成手法之一。
容易得到的芳基卤化物和芳基腙之间的Buchwald-Hartwig偶联合成方法也被开发出来(Buchwald改良)。

- 基本文献
・Fischer, E.; Jourdan, F. Ber. 1883, 16, 2241.
・Fischer, E.; Hess, O. Ber. 1884, 17, 559.
・Miyata, O.; Kimura, Y.; Muroya, K.; Hiramatsu, H.; Naito, T. Tetrahedron Lett. 1997, 62, 425. DOI: 10.1016/S0040-4039(99)00583-3
<Buchwald Modification>
・Wagaw, S.; Yang, B. H.; Buchwald, S. L. J. Am. Chem. Soc. 1998, 120, 6621. DOI: 10.1021/ja981045r
・Wagaw, S.; Yang, B. H.; Buchwald, S. L. J. Am. Chem. Soc. 1999, 121, 10251. DOI: 10.1021/ja992077x
< 综述>
・Van Orden, R. B.; Lindwell, H. G. Chem. Rev. 1942, 30, 69. DOI: 10.1021/cr60095a004
・Robinson, B. Chem. Rev. 1963, 63, 373. DOI: 10.1021/cr60224a003
・Robinson, B. Chem. Rev. 1969, 69, 227. DOI: 10.1021/cr60258a004
・Robinson, B. “The Fischer Indole Synthesis”, John Wiley & Sons Inc., New York, 1982.
・Inman, M.; Moody, C. Chem. Sci. 2012, doi:10.1039/C2SC21185H
- 反应机理

- 反应实例
Peduncularine的合成[1]

Indometacin的合成

Haplophytine的合成[2]

不对称催化的费舍尔吲哚合成[3]

- 实验步骤
- 实验技巧
- 参考文献
[2] Ueda, H.; Satoh, H.; Matsumoto, K.; Sugimoto, K.; Fukuyama, T.; Tokuyama, H. Angew. Chem. Int. Ed. 2009, 48, 7600. doi: 10.1002/anie.200902192
[3] Muller, S.; Webber, M. J.; List, B. J. Am. Chem. Soc. 2011, 133, 18534. DOI: 10.1021/ja2092163
第一代:异腈法
第二代:异硫氰酸酯法

- 概要
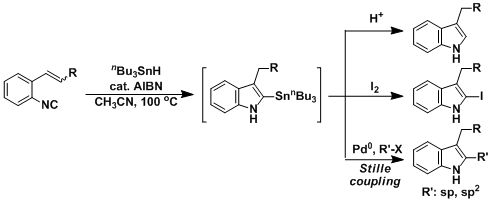
由自由基引发剂及三丁基氢化锡、是合成3-取代和2,3-二取代吲哚的强有力的方法。
特别是第二代方法,是引入多种取代基的有效方法 。在2位的sp3碳原子上引入基团(通过偶联反应通常不能实现)。
原料的硫氰酸酯,可由喹啉的开环反应制备,反应简便易行,并能应用于大量合成。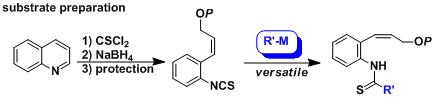
- 基本文献
・Fukuyama, T.; Chen, X.; Peng, G. J. Am. Chem. Soc. 1994, 116, 3127. DOI: 10.1021/ja00086a054
・Tokuyama, H.; Yamashita, T.; Reding, M. T.; Kaburagi, Y.; Fukuyama, T. J. Am. Chem. Soc. 1999,121, 3791. DOI: 10.1021/ja983681v
- 反应机理
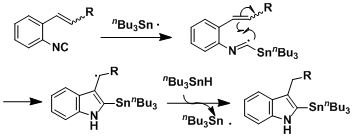
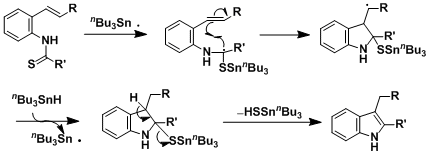
两种方法都是由自由基环化机理进行的。烯烃是反式构型时,热力学稳定的六元环副产物(四氢喹啉)会增加。
- 反应实例
本吲哚合成是合成(+)-Vinblastine中至关重要的步骤[1]、是有機合成化学金字塔中的一个经典实例。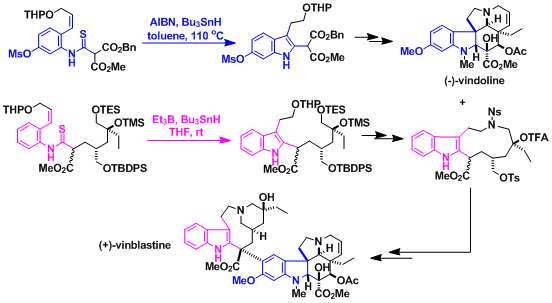
- 实验步骤
- 实验技巧
- 参考文献