作者:石油醚
导读:
南开大学化学学院朱守非课题组报道了首例Brønsted酸催化的不对称Heyns重排反应,以高收率(up to 99%)和高对映选择性(up to 96% ee)合成了系列α-芳基-α-氨基酮,这是此前不对称Heyns反应无法合成的。该反应条件温和,操作简单,收率和选择性高,易于放大,实用性好。该方法被成功用于多种手性胺合成,提高了一些生物活性分子的合成效率。机理实验表明手性磷酸催化了酮胺缩合、亚胺异构化和烯醇质子迁移三个过程,并在催化质子转移过程中实现了手性控制。该成果近期发表于Angew. Chem. Int. Ed. 2022, (https://doi.org/10.1002/anie.202212976),博士生曹晋是文章的第一作者。

图1. Brønsted酸催化高对映选择性Heyns重排反应
“Highly Enantioselective Brønsted Acid Catalyzed Heyns Rearrangement.
Jin Cao, Yu-Xuan Su, Xin-Yu Zhang, Shou-Fei Zhu*
Angew. Chem. Int. Ed. 2022, ASAP. Doi: 10.1002/anie.202212976.
正文:
手性胺是很多天然产物和药物的核心结构单元,也广泛用作手性配体和催化剂。因此,发展从简单原料合成手性胺的方法具有重要意义。Heyns重排反应通过α-羟基酮与胺反应直接合成α-胺基酮,仅产生一分子水为副产物,是合成手性胺的高效方法。近年来,有报道使用手性碱催化剂实现了该反应的不对称转化,但存在催化剂用量高、对映选择性不理想、底物局限等问题。例如,手性碱催化体系仅用于合成手性α-烷基-α-胺基酮,在合成α-芳基-α-胺基酮中未见成功报道。这可能是由于α-芳基-α-胺基酮具有更强酸性的α-H,在碱性催化剂存在下易于消旋化。
此前,南开大学朱守非课题组提出了“手性质子梭催化剂”概念,使用同时含有Brønsted酸和Brønsted碱的手性双功能催化剂作为手性质子梭,通过协同的方式,直接催化烯醇及烯醇盐中间体的质子迁移过程,实现了多种极具挑战的质子转移反应的有效手性控制(Angew. Chem. Int. Ed. 2011, 50, 11483; Angew. Chem. Int. Ed. 2014, 53,3913; Chem. Sci. 2014, 5, 1442; J. Am. Chem. Soc. 2015, 137, 8700; Chem. Sci. 2016, 7, 1104; Chem. Sci. 2017, 8, 7197; Acta Chim. Sinica. 2018, 76, 883; ACS Catal. 2019, 9, 6522; Org. Lett. 2019, 21, 9391; Science 2019, 366, 990; J. Am. Chem. Soc. 2020, 142, 10557; J. Am. Chem. Soc. 2021, 143, 6962; ACS Catal. 2022, 12, 13143)。在上述研究基础上,他们实现了首例手性Brønsted酸催化的不对称Heyns重排反应,以高收率(up to 99%)和高对映选择性(up to 96% ee)合成了系列手性α-芳基-α-胺基酮化合物,这是此前不对称Heyns反应无法合成的。

图2. 对映选择性Heyns重排反应
该反应效率很高。在反应普适性考察中,所有的底物都能在30分钟内完全转化(图3)。反应的底物适用范围较广,α-羟基酮两侧取代基可以为芳基-烷基和芳基-芳基,均能获得优秀的收率和对映选择性。芳基上带有吸电子取代基能给出更好的结果,且产物在反应过程中不会发生消旋化。烷基基团可以是直链烷基、支链烷基、官能化的烷基和苄基。带有给电子基和弱吸电子基的芳胺底物能给出较好的结果,但酰胺和烷基胺在此反应中没有活性。反应可以耐受稠环、芳杂环,以及卤素、酯基、硝基、硅基等多种官能团。

Reaction conditions: 1/2/(R)-10e = 0.1:0.6:0.005 (mmol), in 0.5 mL of solvent at 60 °C, 30 min. [a] Reaction performed in 0.2 mmol scale. [b] (S)-10e was used as catalyst. [c] 1 mL PhOMe was used as solvent.
图3. 底物适用范围
该反应操作简单,容易放大,所得手性α-芳基-α-胺基酮产物可以对映选择性保持地转化为多种手性胺,展现出很好的应用潜力(图4)。作者还将该方法成功应用于生物活性分子(TNF-α抑制剂、脱氧cytoxazone)的合成中,显著提高了合成效率。

(a) BH3-SMe2, THF, -20 °C. (b) RMgBr, THF, -78 °C. (c) Triphosgene, Et3N, THF. (d) nPrMgBr, THF, 0 °C. (e) TiCl4, Ph2CHNH2, Et2O, 0 °C – rt. Then BH3-SMe2, -30 °C. (f) Diethyl azodicarboxylate, PPh3, THF, 0 °C – rt. (g) Ceric ammonium nitrate, MeCN/H2O, 0 °C – rt. (h) Trichloroisocyanuric acid, H2SO4, MeCN/H2O, rt.
图4. 克级规模实验及产物转化
该Brønsted酸催化的不对称Heyns重排存在两种可能的反应过程(图5A)。作者通过氘代实验排除了1,2-负氢迁移的机制(图5B),说明了反应中可能经历了烯醇中间体并发生了氢氘交换(图5C);α-羟基酮底物的光学纯度不影响反应的对映选择性(图5D),进一步排除了负氢迁移的可能性;KIE实验表明烯醇的生成不是反应的决速步(图5 E和F)。作者认为,手性磷酸催化了酮胺缩合、亚胺异构化和烯醇质子迁移三个过程,并在催化质子转移过程中实现了手性控制。
图5. 机理实验
理论计算表明过渡态能量与两方面因素密切相关:一是催化剂与烯醇的轨道重叠程度,二是烯醇的稳定性。在优势过渡态TS-R中,分子轨道重叠优于相应的TS-S过渡态,作者认为过渡态轨道最大重叠需求是手性质子梭实现手性控制的主要驱动力。此外,作者观察到了底物芳环取代基电性显著影响对映选择性的现象。理论计算表明,缺电子芳环与烯醇的二面角发生了更大程度的扭转。这种形变影响了底物与催化剂的相互作用,最终表现为底物电性对于反应对映选择性的影响。(图6)

图6. DFT计算
总之,朱守非课题组实现了首例手性Brønsted酸催化的不对称Heyns重排反应。该反应效率高,底物适用范围广,能在温和条件下高对映选择性得到手性中心具有较强酸性氢的手性α-芳基-α-胺基酮化合物,这是文献中手性碱催化体系所无法实现的。反应产物可以发生丰富的转化,为复杂含氮功能分子的合成带来新的机遇。机理实验表明,反应经历了亚胺到烯胺以及烯醇到酮式两次异构化过程。手性磷酸通过氢键网络促进了烯醇/胺中间体的不对称质子转移。该反应为不对称Heyns重排反应的实际应用带来可能,其新颖的催化体系也为经历活性烯醇/胺中间体的反应的研究带来新的思路。
(朱守非教授供稿)
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载
作者开发了一种使用手性路易斯酸催化剂的新型环丁酮不对称合成方法。实现了高非对映体/对映体选择的多米诺型的环丙烷化→半频哪醇重排形成手性多取代环丁酮。
催化不对称环丁酮合成
环丁烷衍生物是药物和天然产物中常见的骨架。特别是,环丁酮是一种非常有用的结构单元,可以通过利用环应变的开环和环膨胀反应合成为各种化合物,最近已经有几篇针对该骨架的催化不对称合成方法的报道。例如、最近Dong等人报道了使用钴催化的分子内不对称加氢酰化合成环丁酮(图1A)[1]。另一方面,通过催化半环醇重排环丙醇衍生物合成环丁酮的方法也已经被报道。Alexakis等人在手性磷酸(CPA)催化剂存在下通过链烯基环丙醇的半微量醇重排开发出了β-卤代螺环丁酮合成法(图1B)[2]。作为使用不对称过渡金属催化剂的实例,Toste与Trost等人分别报道了使用金或钯作为催化剂的链烯基环丙醇的半频哪醇重排(图1C)[3]。此外,还报道了通过酸,碱和热将半乳糖醇重排成羰基部分合成环丁酮,但没有对映选择性实例(图1D)[4]。
最近,Ryu教授及其同事使用手性氧杂硼杂环鎓离子(COBI)作为路易斯酸催化剂,通过α-甲硅烷氧基丙烯醛和重氮酯的反应进行连续环丙烷化/半频哪醇重排, 成功地合成了具有立体/对映选择性邻近手性中心的环丁酮(图1E),今天小编就带大家来看一下这个反应。

图1.催化不对称环丁酮的合成
“Asymmetric Synthesis of Cyclobutanone via Lewis Acid Catalyzed Tandem Cyclopropanation/Semipinacol Rearrangement”
Shim, S. Y.; Choi, Y.; Ryu, D. H. J. Am. Chem. Soc.2018, 140, 11184−11188. DOI: 10.1021/jacs.8b06835
论文作者介绍

研究者:Do Hyun Ryu
经历:
1993-1997 Ph. D., Department of Chemistry, KAIST (Prof. Sung Ho Kang)
1997-2001 Associate Research Scientist, SK Chemicals, Life Science Institute
2000-2002 Postdoc Fellow, Harvard Medical School (Prof. Robert R. Rando)
2002-2005 Postdoc Fellow, Harvard University (Prof. E. J. Corey)
2005-2009Assistant Professor, Sungkyunkwan University
2009-2015Associate Professor, Sungkyunkwan University
2015- Professor, Sungkyunkwan University
研究内容:催化剂开发、不对称反应开发、全合成研究、化学生物学
论文概要
作者在先前的研究中报道了使用COBI催化剂对不饱和醛和重氮酯进行不对称环丙烷化[5]。在这次研究中,作者使用α-硅氧基丙烯醛,并且利用同样的手法如果能先生成1-甲酰基-1-甲硅烷氧基环丙烷1、然后发生半频哪醇重排,最后形成环丁酮2。反应实际上按预期进行,并且可以应用于各种重氮酯(图2A)。即使使用具有卤素或氰基等官能团的芳基,烷基或具有大体积酯的重氮酯,反应也以高收率,高非对映体/对映体选择性进行。
根据各种对照实验的结果,作者提出了通过过渡态4产生环丁酮2的机理(图2B)。首先,COBI催化剂与不饱和醛的羰基部分配位,重氮酯的1,4-加成平面选择性地发生,并且通过随后的环丙烷化形成4。此外,对由COBI催化剂活化的羰基部分发生半频哪醇重排,并且通过甲硅烷基转移获得目标环丁酮2。
因此,这篇文章报道了一种合成具有连续不对称中心的环丁酮的新方法。 将来,可以预期该方法可用作各种有用复杂化合物中间体合成。

图2. (A)底物适用范围、(B)过渡态模型
参考文献
- Kim, D. K.; Riedel, J.; Kim, R. S.; Dong, V. M. J. Am. Chem. Soc. 2017, 139, 10208. DOI:10.1021/jacs.7b05327
- Romanov-Michailidis,F.;Gueńeé,L.;Alexakis, Angew. Chem., Int. Ed. 2013, 52, 9266. DOI: 10.1002/anie.201303527
- [a]Kleinbeck, F.; Toste, F. D. J. Am. Chem. Soc. 2009, 131, 9178. DOI: 10.1021/ja904055z[b] Trost, B. M.; Yasukata, T.J. Am. Chem. Soc.2001, 123, 7162. DOI: 10.1021/ja010504c
- [a]Paukstelis, J. V.; Kao, J. L. J. Am. Chem. Soc. 1972, 94, 4783. 10.1021/ja00768a086[b] Appendino, G.; Bertolino, A.; Minassi, A.; Annunziata, R.; Szallasi, A.; de Petrocellis, L.; Di Marzo, V. Eur.J. Org. Chem. 2004, 2004, 3413. DOI: 10.1002/ejoc.200400122
- Gao, L.; Hwang, G.-S.; Ryu, D. H. J. Am. Chem. Soc. 2011,133, 20708. DOI: 10.1021/ja209270e
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!
概要
β-酮酸烯丙酯发生Claisen重排与脱羧反应,得到γ,δ-不饱和酮。
在Pd或者Ru催化的条件下,该反应能在温和的条件下进行,并且经常也被用于不对称中心的构建。丙二酸烯丙基酯也能作为该反应的底物。
基本文献
- Carroll, M. F. J. Chem. Soc. 1940, 704. DOI: 10.1039/JR9400000704
- Carroll, M. F. J. Chem. Soc. 1941, 507. DOI: 10.1039/JR9410000507
- Kimel, K; Cope, A. C. J. Am. Chem. Soc. 1943, 65, 1992. DOI: 10.1021/ja01250a049
- Wilson, A. R.; Augelli, C. E. Org. Synth. 1990, 68, 210.
- Castro, A. M. M. Chem. Rev. 2004, 104, 2939. DOI: 10.1021/cr020703u
<metal-catalyzed conditions>
- Shimizu, I.; Yamada, T.; Tsuji, J. Tetrahedron Lett. 1980, 21, 3199. DOI: 10.1016/S0040-4039(00)77444-2
- Burger, E. C.; Tunge, J. A. Org. Lett. 2004, 6, 4113. doi:10.1021/ol048149t
- You, S.-L. Dai, L.-X. Angew. Chem. Int. Ed. 2006, 45, 5246. doi:10.1002/anie.200601889
- Austeri, M.; Buron, F.; Constant, S.; Lacour, J. Linder, D.; Muller, J.; Tortoioli, S. Pure Appl. Chem.2008, 80, 967. doi:10.1351/pac200880050967
- Hong, A.Y.; Stoltz, B. M. Eur. J. Org. Chem. 2013, 14, 2745. DOI: 10.1002/ejoc.201201761
反应机理

反应实例
Stoltz等人通过不懈的研究开发了用手性Pd催化剂构筑手性四级碳中心的方法。该方法可以应用于多种类Cyanthiwigin的全合成。[1]

实验步骤
实验技巧
参考文献
[1] (a) Enquist, J. A.; Stoltz, B. M. Nature 2008, 453, 1228. doi:10.1038/nature07046 (b) Enquist, J. A. Jr.; Virgil, S. C.; Stoltz, B. M. Chem. Eur. J. 2011, 17, 9957. DOI: 10.1002/chem.201100425- Sorgi, K. L., Scott, L., Maryanoff, C. A. Tetrahedron Lett. 1995, 36, 3597. DOI: 10.1016/0040-4039(95)00602-9
- Enders, D., Knopp, M. Tetrahedron 1996, 52, 5805. DOI: 10.1016/0040-4020(96)00236-0
- Sobenina, L. N., Mikhaleva, A. I., Petrova, O. V., Polovnikova, R. I., Trofimov, B. A. Unknown pathway of the Carroll reaction. Russ. J. Org. Chem. 1997, 33, 1041
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!
概要
O-芳基亚胺酯经过重排转化成芳香胺的反应。
基本文献
- Mumm, O. et al. Ber. 1915, 48, 379.
- Chapman, A, W. J. Chem. Soc. 1925, 127, 1992.
- Chapman, A. W. J. Chem. Soc. 1927, 129, 174.
- Chapman, A. W. J. Chem. Soc. 1929, 131, 569.
- Schulenberg, J. W. et al. Org. React. 1965, 14, 1.
反应机理
(参考:J. Chem. Soc. Perkin Trans. 2 1983, 1369.)

反应实例
实验步骤
实验技巧
参考文献
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!
- 概要
硅烯醇醚在mCPBA或二环氧乙烷中被环氧化后,能够快速进行重排,得到α-硅氧基酮。该反应经常被用于在α位选择性导入羟基。
- 基本文献
・ Rubottom, G. M.; Vazquez, M. A.; Pelegrina, D. R. Tetrahedron Lett. 1974, 15, 4319. doi:10.1016/S0040-4039(01)92153-7
・ Brook, A. G.; Macrae, D. M. J. Organomet. Chem. 1974, 77, C19. doi:10.1016/S0022-328X(00)81332-7
・ Hassner, A.; Reuss, R. H.; Pinnick, H. W. J. Org. Chem. 1975, 40, 3427. DOI: 10.1021/jo00911a027
- 反应机理

- 反应实例
(-)-Stenine的合成[1]

- 实验步骤
- 实验技巧
- 参考文献
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载
- 概要
对于低级羰基碳正离子中心,邻位的碳发生1,2重排,伴有氢,烷基,芳基迁移,得到更加稳定的碳正离子的重排反应。
一般来说,相比于H原子、炔基、烷基来说,芳基,烯基更容易发生重排。另外,拥有供电子基团的最容易进行重排。
- 基本文献
・Wagner, G. J. Russ. Phys. Chem. Soc. 1899, 31, 690.
・Meerwein, H. Ann. 1914, 405, 129. doi:10.1002/jlac.19144050202
・Cargill, R. L.; Jackson, T. E.; Peet, N. P.; Pond, D. M. Acc. Chem. Res. 1974, 7, 106. DOI: 10.1021/ar50076a002
・Olah, G. A. Acc. Chem. Res. 1976, 9, 41. DOI: 10.1021/ar50098a001
・Hogeveen, H.; Van Krutchten, E. M. G. A. Top. Curr. Chem. 1979, 80, 89.
・Hanson, J. R. Comprehensive Organic Synthesis 1991, 3, 705-719.
・Birladeanu, L. J. Chem. Edu. 2000, 77, 858.
- 反应机理
如果重排前的碳正离子比较稳定的话,首先会发生键的旋转,然后再发生重排。所以经常最后会得到立体混合物。

- 反应实例
- 实验步骤
- 实验技巧
- 参考文献
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载
- 概要
在路易斯酸作用下,芬酯化合物的酰基发生的重排反应。生成产物的邻/对位的选择性的话,是根据反应条件所决定的。低温(100℃以下) 的话,主要生成对位产物,而高温的话邻位是主要产物。该反应可以视作分子内Friedel-Crafts酰化的一种。
紫外线照射同样能诱发该重排反应(被称为光Fries重排)。
- 基本文献
・Fries, K.; Fink, G. Ber. 1908, 41, 4271. doi:10.1002/cber.190804103146
・Fries, K.; Pfaffendorf, W. Ber. 1910, 43, 212. doi:10.1002/cber.19100430131
・Blatt, A.H. Org. React. 1942, 1, 342.
・Effenberger, F.; Klenk, H.; Reiter, P. L. Angew. Chem. Int. Ed., Engl. 1973, 12, 775. DOI: 10.1002/anie.197307751
- 反应机理
酸性条件下,反应循环是通过形成酰基阳离子进行的,光条件下的话,是按照自由基的开裂,再结合的过程。

- 反应实例
Diazonamide core的合成研究[1]

- 实验步骤
- 实验技巧
- 参考文献
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载
- 概要
以酸或者酰卤作为反应底物,引发形成的酰基叠氮中间体在加热条件下重排,形成异氰酸酯。如果此时,有水介入反应的话,异氰酸酯会立即加水分解,形成脱掉一个碳的胺。此外,该方法是为数不多的、能够合成具有光学活性的链状胺的方法之一。
除了水以外,如果使用一些适当的醇作为反应剂的话,,就可以很方便的形成各种氨基酸酯类基团作为胺基的保护基,比如Boc、Cbz等。因此,此反应是应用性极其高的一个重排反应。
叠氮钠具有一定的爆炸危险性,一般的话,我们用叠氮磷酸二苯酯(DPPA)来代替其使用。用DPPA还有个好处就是,酸可以直接在比较温和的条件下形成酰基叠氮中间体,然后进行库尔提斯重排反应。
- 基本文献
・Curtius, T. Ber. 1890, 23, 3023.
・Curtius, T. J. Prakt. Chem. 1894, 50, 275.
・Shioiri, T.; Ninomiya, K.; Yamada, S.-i. J. Am. Chem. Soc. 1972, 94, 6203. DOI: 10.1021/ja00772a052
・Ninomiya, K.; Shioiri, T.; Yamada, S.-i. Tetrahedron 1974, 30, 2151. doi:10.1016/S0040-4020(01)97352-1
・Smith, P. A. S. Org. React. 1946, 3, 337.
・Scriven, E. F.; Turnbull, K.Chem. Rev. 1988, 88, 297. DOI: 10.1021/cr00084a001
・Wolff, O.; Waldvogel, S. R. Synthesis 2004, 1303. DOI: 10.1055/s-2004-815965
- 反应机理
氮宾(nitrene)式的中间体形成后,保持立体构型不变进行重排反应。当R=H的情况下,后续的脱碳酸化反应后形成了胺。
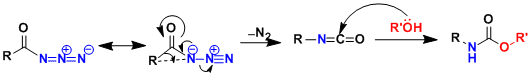
- 反应实例
DPPA参与的反应。[1]

对于α-叔胺等合成比较困难的结构运用此条件也能够很有效的合成。 [2]
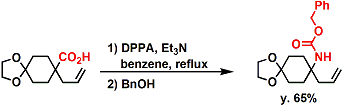
奥司他韦(Tamiflu)的合成。 [3]
- 实验步骤
- 实验技巧
- 参考文献
[2] Murashige, K. et al. Tetrahedron 2002, 58, 4917. doi:10.1016/S0040-4020(02)00436-2
[3] Yamatsugu, K.; Yin, L.; Kamijo, S.; Kimura, Y.; Kanai, M.; Shibasaki, M. Angew. Chem., Intl. Ed. 2009, 48, 1070. DOI: 10.1002/anie.200804777